Le cancer est en France un problème de santé publique majeur. La Commission d’orientation sur le cancer, dans un rapport de janvier 2003, fait état de 278 000 nouveaux cas et 150 000 décès dus à cette maladie pour l’année 2000. Les cancers représentent globalement la deuxième cause de mortalité en France derrière les affections cardiovasculaires (28 % des décès). C’est la première cause de mortalité chez l’homme (32 %) et la seconde chez la femme (23 %). L’environnement physique ou chimique, naturel ou synthétique, domestique, urbain, rural ou professionnel est depuis longtemps soupçonné d’être impliqué dans la production de cancers en association avec les caractéristiques héréditaires des individus et dans des circonstances favorisantes. S’agissant de pathologies plurifactorielles, il est difficile d’évaluer précisément la part des cancers ayant une origine professionnelle, néanmoins une fourchette de 4 à 8,5 % des cas est retenue dans plusieurs rapports.
La transformation d’une cellule normale en cellule cancéreuse résulte de l’accumulation d’altérations génétiques modifiant l’activité de gènes bien spécifiques. Ainsi, la transformation néoplasique comprend plusieurs étapes : activation d’un proto-oncogène, inactivation d’un gène suppresseur de tumeur et dysfonctionnement d’un point de contrôle du cycle cellulaire. Le cancer est une pathologie à point de départ précoce avec des effets différés dans le temps. La relation entre génotoxicité (effet d’agents qui interagissent avec l’ADN et/ou la machinerie cellulaire qui maintient l’intégrité du génome), mutagenèse et cancérogenèse est admise mais sa démonstration demeure malaisée en raison de l’intrication de mécanismes stochastiques et déterministes, de facteurs héréditaires et de phénomènes acquis.
Mutations et cancers
L’ADN dans tous ses états
Il est bien établi que l’ADN représente le support moléculaire biologique porteur de l’information génétique de la plupart des êtres vivants, à l’exception notable des virus à ARN. L’ADN est une macromolécule biologique à laquelle sont attribuées les structures primaire, secondaire et tertiaire. Il apparaît que la séquence en nucléotides (structure primaire), la configuration et la situation des nucléotides au sein de la double hélice (structure secondaire) et l’état sous lequel se trouve la molécule d’ADN (en cours de réplication ou de transcription, sous forme de chromatine ou de chromosome : structure tertiaire) jouent un rôle considérable dans la survenue des lésions puis des mutations.
Mutagenèse et cancérogenèse
La molécule d’ADN est une structure dynamique sujette à de constants changements. Ces variations sont consécutives, d’une part à des erreurs spontanées, d’autre part à des lésions de l’ADN induites par des agents physiques ou chimiques (UV, radiations ionisantes, produits chimiques) qualifiés de génotoxiques. La plupart de ces dommages génétiques sont réparés et sans conséquence (réparation ad integrum). Quelquefois, si le dommage est trop important, les capacités de réparation de la cellule sont dépassées et la cellule peut mourir (par exemple par apoptose). Cependant, des agressions répétées sur le génome augmentent la probabilité de survenue de lésions irréversibles (mutations) et peuvent être à l’origine de l’initiation d’un processus cancérogène (Figure 1).
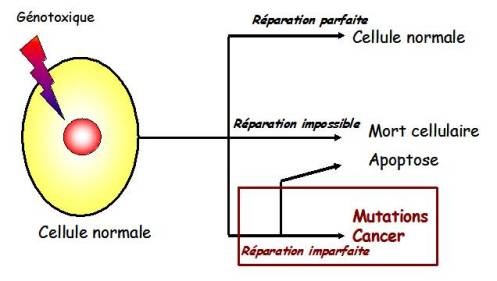
Cancer = Environnement + Hérédité + Circonstances favorisantes
La transformation d’une cellule normale en une cellule cancéreuse requiert plusieurs mutations spécifiques (le chiffre de six est souvent cité). Si le taux typique de mutations dans une cellule est de l’ordre de 10-6 par mitose et par gène (après réparation des mésappariements), la probabilité pour chaque gène de muter, dans les 1014 cellules qui constituent notre organisme, au cours des 1016 divisions cellulaires qui ont lieu au cours de notre vie entière est de : 10-6 x 1016 soit 1010 occasions de muter. Cette probabilité augmente s’il existe un déficit héréditaire de réparation des mésappariements, ainsi certains syndromes d’instabilité génétique sont associés à un accroissement sévère du risque de cancer (exemple du xeroderma pigmentosum associant une hypersensibilité aux UV du fait d’une incapacité de l’ADN à réparer les lésions secondaires au rayonnement solaire et des cancers cutanés multiples).
Un exemple de test de génotoxicité : le test des micronoyaux
Intérêt
Dans le cadre des relations santé - environnement, le domaine de la toxicologie génétique s’est révélé être essentiel puisque la détermination des facteurs environnementaux susceptibles d’interagir, directement ou non, avec le patrimoine génétique des cellules est devenue nécessaire dans le cadre de la prévention du risque cancérogène. Compte tenu de la grande diversité des anomalies susceptibles d’être induites au niveau d’un patrimoine génétique, il n’existe pas un, mais plusieurs tests de mutagenèse susceptibles de révéler tels ou tels types de lésions ou de mutations. Le choix des tests de toxicologie génétique doit se faire en fonction du mode d’action d’un agent génotoxique : par exemple le caractère aneugène (induisant des pertes de chromosomes) d’une substance est évalué au mieux par le test des micronoyaux (MN). Le test des MN appliqué à la biosurveillance du risque cancérogène professionnel présente l’intérêt de pouvoir alerter le préventeur de la pénétration de génotoxiques dans l’organisme et/ou de l’existence de modifications biologiques précoces, contemporaines de l’exposition, consécutives à des interactions avec le matériel génétique, moment où la probabilité de progression tumorale est encore faible mais où l’efficacité de l’intervention est maximale (Figure 2).
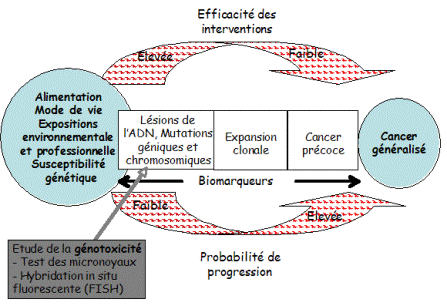
Test des micronoyaux
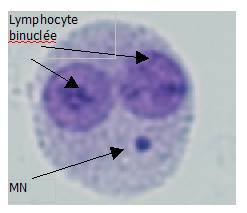
Les MN, également appelés corps de Howell-Jolly par les hématologistes, peuvent être, soit le témoin d’une instabilité génétique, soit un biomarqueur d’effet mettant en évidence des dommages chromosomiques induits par des agents mutagènes/cancérogènes. La technique des MN sur lymphocytes binucléés en culture (qui a été initialement décrite par Fenech et Morley en 1985) consiste à bloquer la cytodiérèse par ajout de cytochalasine B à la 44ème heure de culture (Figure 3). Les MN ont pour origine des fragments chromosomiques ou des chromosomes entiers qui ne migrent pas lors de l’anaphase. Ces deux types de contenu des MN correspondent à des mécanismes de formation fondamentalement différents.
Hybridation in situ fluorescente (FISH)
L’association de l’hybridation in situ fluorescente permet de différencier les MN contenant des fragments chromosomiques acentromériques (cassures chromosomiques) et ceux contenant des chromosomes entiers centromériques (pertes chromosomiques) consécutifs respectivement à des évènements clastogènes ou aneugènes. Elle permet d’identifier la région centromérique de chaque chromosome humain, sur des préparations chromosomiques en métaphase. Pour chaque cellule micronucléée, le nombre de MN est enregistré de même que la présence ou non de centromères dans les MN et le nombre de spots de fluorescence (correspondant chacun à un centromère donc à un chromosome) au sein de chaque MN (Figure 4).
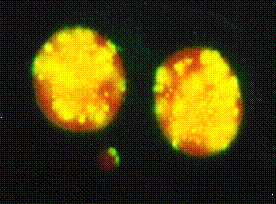
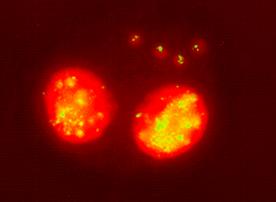
Le MN est un biomarqueur intégrant de nombreux facteurs de variation (sexe, âge, tabagisme, alcool, habitudes alimentaires). Il est par conséquent intéressant de rechercher un sous type de MN centromériques (perte de plusieurs chromosomes et anomalies de duplication du centrosome, centre organisateur) dont la significativité et la prédictivité du risque cancérogène seraient plus importantes.
Susceptibilités génétiques
Polymorphisme génétique
Le polymorphisme génétique correspond à l’existence de variations dans la séquence de l’ADN à un locus donné. Ces variations sont susceptibles de moduler l’activité des enzymes impliquées dans le métabolisme des xénobiotiques (enzymes de phase I responsable de phénomènes de toxification comme les cytochromes P450 mono-oxygénases, ou enzymes de phase II responsables de phénomènes de détoxification/conjugaison comme les glutathion S-transférases, N-acétyl-transférases, sulfotransférases) ou dans la réparation des lésions de l’ADN (tels que hOGG1, XRCC1 ou XRCC3), mais n’ont pas de conséquences pathologiques du fait d’un dysfonction du gène ou de la protéine, ce qui les différencie des mutations à proprement parler.
Altérations des capacités de réparation de l’ADN avec l’âge
A chacune des étapes de la cancérogenèse, depuis la plus précoce (la génotoxicité) jusqu’à la plus tardive (le clone tumoral constitué), l’environnement et l’hérédité sont en étroite interaction. Les agents mutagènes/cancérogènes subissent, en général, plusieurs transformations métaboliques dans l’organisme, ce qui peut conduire à leur élimination mais aussi parfois à la formation de composés capables d’altérer les macromolécules cellulaires et pouvant faire l’objet d’une réparation parfaite ou fautive (Figure 5).
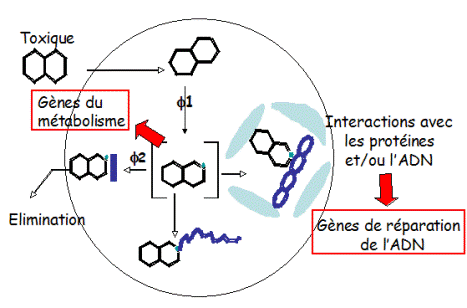
Plusieurs études s’intéressant aux interactions gènes/environnement ont porté sur les polymorphismes des gènes du métabolisme et l’exposition professionnelle à divers agents tels que les amines aromatiques ou les hydrocarbures aromatiques polycycliques. Il a pu être ainsi mis en évidence un effet conjoint de l’exposition aux amines aromatiques et du polymorphisme des N-acétyl-transférases (NAT2) sur le risque de cancer de la vessie (risque relatif plus élevé si le sujet est acétyleur lent). De plus, les mécanismes de réparation deviennent de moins en moins efficaces avec l’âge et cette altération des capacités de réparation des lésions de l’ADN avec le temps peut expliquer l’accroissement vraisemblable de la sensibilité aux agents mutagènes/cancérogènes.
Conclusion
Du fait de sa capacité à détecter les effets précoces d’agents mutagènes/cancérogènes, à identifier les mutations somatiques et à prédire le risque cancérogène, le test des MN associé à la prise en compte des susceptibilités génétiques devrait prendre de plus en plus d’importance dans le futur. L’établissement d’un lien de causalité entre l’exposition à un environnement et la production d’un cancer est extrêmement difficile à établir en raison notamment du temps de latence de l’apparition de la maladie par opposition à des incertitudes sur un seuil d’action cancérogène (effets des très faibles doses répétées ?), à l’implication forte de l’hérédité et de phénomènes associés acquis, et à des interactions multiples entre les divers composants de l’environnement auquel un individu est exposé au cours de sa vie personnelle et professionnelle.
Bibliographie
KIRSCH-VOLDERS M., De Boeck M., LISON D., « Génotoxicité et activité professionnelle », in Encyl Méd Chir (Editions Scientifiques et Médicales Elsevier SAS, Paris), Toxicologie-Pathologie professionnelle, 2002.
MATEUCA R., LOMBAERT N., AKA P.V., Decordier I., Kirsch-Volders M., "Chromosomal changes: induction, detection methods and applicability in human biomonitoring", in Biochimie, 2006, doi:10.1016/j.biochi.2006.07.004 (sous presse).
IARMARCOVAI G., BOTTA A., ORSIERE T., "Number of centromeric signals in micronuclei and mechanisms of aneuploidy", in Toxicol Lett, n° 166, 2006, pp. 1-10.
BONASSI S., ZNAOR A., CEPPI M., LANDO C., CHANG W.P., HOLLAND N., et al, "An increased micronucleus frequency in peripheral blood lymphocytes predicts the risk of cancer in humans", Carcinogenesis, n° 2006. doi:10.1093/carcin/bgl177 (sous presse).
« Susceptibilités génétiques et expositions professionnelles », in Documents pour le médecin du travail, INRS, n° 83, 2000, 249-258.
GORBUNOVA V., SELUANOV A., "Making ends meet in old age: DSB repair and aging", in Mech Ageing Dev, n°126, 2005, pp. 621-628.
Liens internet :
Textes d’Alain Botta, Thierry Orsière et Jean-Louis Bergé-Lefranc